
Authors: Igor Sokolov, Lucia Garbini
Imagine you could predict how molecules behave, how bonds break and form, how materials conduct or insulate, without tracking every single electron. That’s essentially what modern computational chemistry does: instead of following each electron individually, scientists model the overall “cloud” of electron density. This simplification is powerful enough to run on computers, which is why it underpins everything from drug design to battery research.
But there’s a catch. One key piece of the model, the exchange‑correlation (XC) functional, has to capture how electrons avoid each other and form chemical bonds. Nobody knows its exact mathematical form, so chemists rely on a collection of approximations. These approximations work well for many systems, but they often fail for complex cases like strongly correlated materials, reaction barriers, or systems where bonds break and reform.
Pasqal releases QEX, an open‑source library that uses machine learning, and, in the quantum‑enhanced case, quantum computations , to learn better approximations of this functional directly from data.
Grounded in our peer‑reviewed Physical Review A paper, co-authored with Schrödinger, QEX makes it practical for researchers to train and test quantum‑enhanced XC functionals inside a full Kohn‑Sham Density Functional Theory (KS‑DFT) workflow, end‑to‑end.
A smarter way to guess the XC functional
In most simulations, chemists plug in a standard approximation for the XC functional and hope that it works well enough for the system in question. Over the years, these approximations have improved, but they still behave like a ladder of compromises: each step adds more information about the electron density at the cost of new failure modes. No universal approximation exists.
QEX shifts the paradigm: instead of guessing a mathematical formula, we treat the XC functional as something that can be learned from data. The idea is to train a model, classical or quantum‑enhanced, that maps the electron density to the XC energy, and then embed that model directly into the self‑consistent KS‑DFT loop. This lets the model learn how electrons correlate, constrained by the physics of the system and the quality of the reference data.
Where quantum‑enhanced XC can help
Classical neural networks are powerful function approximators, but they still work in a compressed, classical feature space. The XC functional takes as input the electron density, a 3D “cloud” of where electrons are likely to be and must implicitly encode complex quantum correlations that grow quickly with the number of electrons.
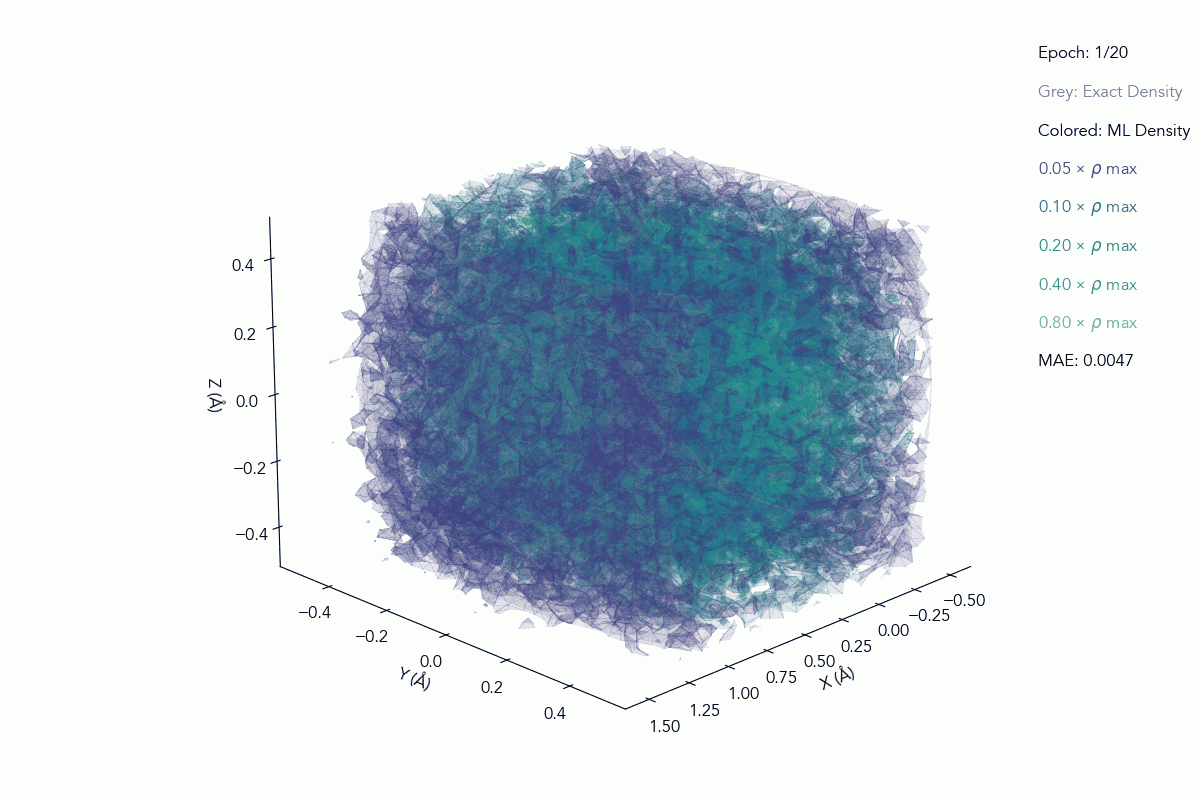
In QEX, the quantum‑enhanced XC functional is realized via digital quantum computations, offering a different way to encode these correlations in the XC term. We don’t treat this as a guaranteed shortcut to higher accuracy; instead, we treat it as a different inductive bias, a different way of encoding structure into the model. QEX lets us test whether quantum‑enhanced XC functionals can generalize better across systems, under the same training conditions as classical networks.
Why this matters for chemistry and materials
The most promising future targets for QEX are strongly correlated materials, transition‑metal oxides, Mott insulators, and high‑Tc superconductors, where standard XC approximations systematically fail. These systems might exhibit multi‑reference character that quantum‑enhanced XC functionals are structurally well‑suited to capture.
Beyond this core use case, better XC accuracy improves many areas where DFT is a bottleneck:
- Materials discovery: more trustworthy energetics for novel materials with tailored electronic or optical properties.
- Energy research: more accurate models of battery electrolytes, catalytic surfaces, and photovoltaic materials.
- Drug design: more reliable predictions of binding energies, reaction barriers, and conformational rankings.
Right now, quantum hardware limits how large and complex the quantum‑enhanced XC models can be, but the framework in QEX is scalable. As neutral‑atom quantum processors grow in size and fidelity, it will become possible to train on more diverse and chemically relevant systems, potentially uncovering new XC models, much as happened in deep learning when larger datasets and models revealed new capabilities.
What researchers can do with QEX
QEX is designed to be a practical, open tool for the community, not just a proof‑of‑concept. With QEX, researchers can:
- Train neural XC functionals end‑to‑end through a fully differentiable KS‑DFT pipeline, for both 1D model systems and 3D molecules.
- Integrate quantum‑enhanced XC functionals that leverage quantum computations on neutral‑atom devices, using different parameterized quantum representations of the XC functional.
- Run controlled comparisons between classical and quantum‑enhanced XC models, studying how representation structure and how the density is encoded affect performance.
- Emulate realistic hardware noise to test how robust quantum‑enhanced XC functionals are to near‑term processor imperfections.
We especially encourage the community to explore classically-challenging molecular systems and, in the future, train XC functionals on quantum hardware , which will be key to understanding where quantum‑enhanced XC truly adds value.
How QEX works in practice
QEX is built on the JAX ecosystem and extends differentiable chemistry frameworks such as JAX‑DFT (for 1D model systems) and PySCFAD (for 3D molecules). The workflow is simple in concept:
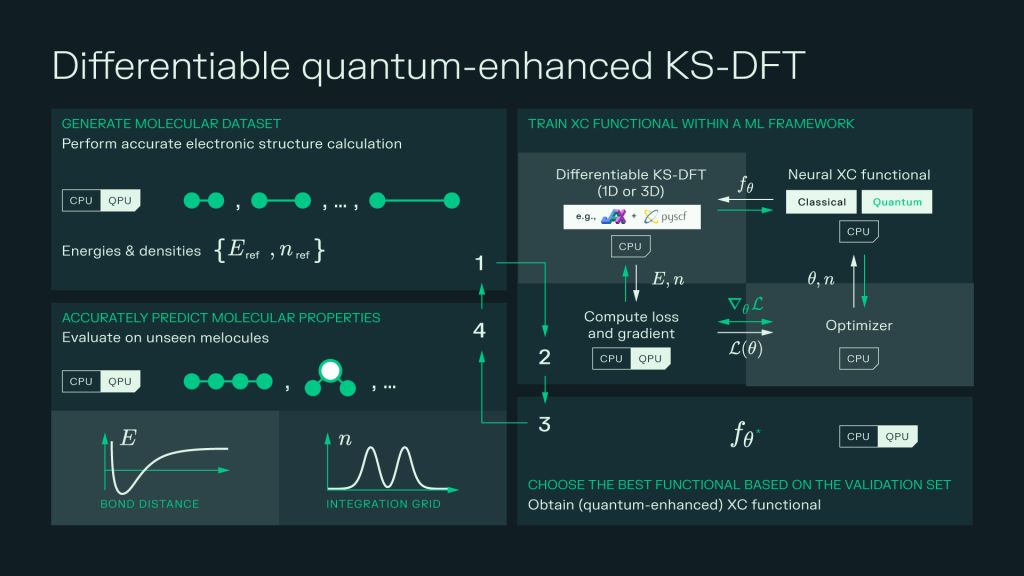
- Generate reference data from high‑accuracy methods like Coupled Cluster Singles, Doubles, and perturbative Triples (CCSD(T)), Density Matrix Renormalization Group (DMRG), or other advanced solvers.
- Define a neural XC functional, a classical neural network or a quantum‑enhanced XC model and plug it into the Kohn‑Sham DFT equations.
- Train the model end‑to‑end: the framework runs the self‑consistent field (SCF) loop, and JAX automatically propagates gradients through the full cycle, updating the neural XC functional to match the reference data.
- Validate on unseen systems and then deploy the trained functional on larger or more complex systems, since the functional acts on density inputs rather than on a fixed number of atoms.
Quantum computations on neutral‑atom devices are integrated via Qadence and Horqrux , which provide differentiable, GPU‑accelerated emulation. This means researchers can experiment with quantum‑enhanced XC functionals today, even before large‑scale fault‑tolerant quantum hardware exists.
What QEX can already do
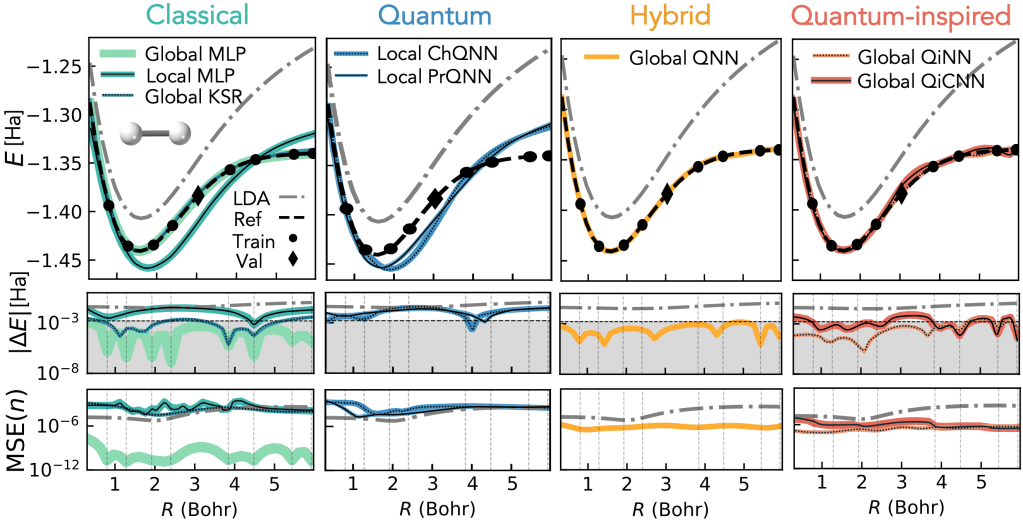
In our Physical Review A paper, we show that QEX‑based quantum‑enhanced XC functionals achieve chemical precision on small hydrogen systems such as H₂, H₄, and the H₂–H₂ dimer, compared with accurate DMRG and FCI/6‑31G reference data. Energy deviations stay below about 1 mHa across the full potential energy surface, including the strongly correlated bond‑stretching regime where standard approximations like LDA and B3LYP fail.
Even more interesting is generalization: a functional trained only on H₂ and H₄ can be evaluated on the H₂–H₂ dimer without retraining and still achieve chemicalprecision (<1 kcal/mol) in large parts of the energy profile. The learned functional captures transferable physics with only a few variational parameters.
Different quantum‑enhanced XC representations, implemented via digital quantum computations, are assessed alongside classical neural networks and achieve competitive accuracy with a similar number of variational parameters. This shows that quantum‑enhanced XC is not only conceptually attractive but also practically competitive.
A subtler but important ingredient is fractional orbital occupation during the SCF cycle. Without fractional occupation, the SCF can oscillate and gradient‑based training can diverge, especially when trying to match exact reference data. Allowing fractional occupations stabilizes the training, letting the model learn the correct physics without getting stuck in unhelpful cycles.
Outlook: foundations, bottlenecks, and next steps
Our work establishes that quantum‑enhanced XC functionals are viable, trainable, and size‑independent, meaning the same functional can be trained on small systems and then evaluated on larger ones without retraining. The quantum‑classical hybrid representation in QEX “slides” over the electron density, so the same functional structure can be reused across systems of different sizes.
However, several bottlenecks remain:
- Architectural: the current ansatz is size‑independent but not automatically chemically transferable.
- Data: training so far is restricted to hydrogen systems; broader transferability will require larger and more chemically diverse reference datasets.
- Hardware: the main bottleneck is training (backpropagation over emulated quantum computations), not inference.
- Conceptual: quantum‑enhanced XC here is motivated as a different inductive bias, not a proof of quantum advantage.
QEX is intentionally open and extensible so that the community can address these challenges together.
Get started and contribute
QEX is available on GitHub under an open‑source license, with source code, documentation, and examples.
Documentation and tutorials help users build and train their own neural XC functionals, classical or quantum‑enhanced. We welcome bug reports, feature requests, and contributions via the issue tracker and contribution guidelines.
If you use QEX in your research, we kindly ask that you cite our Physical Review A article.

